Course:MEDG550/Student Activities/Wolf Hirschhorn Syndrome
What is Wolf Hirschhorn syndrome?
Wolf Hirschhorn (WHS) is a microdeletion syndrome caused by missing genetic material on chromosome 4. This syndrome was first described by Wolf and Hirschhorn in 1965, who both described a condition associated with a deletion of the short arm of chromosome 4 (4p)[1]. The short arm of a chromosome is called the 'p' arm, therefore the syndrome is also known as 4p deletion syndrome. This condition has also previously been described as Pitt-Rogers-Danks syndrome (PRDS) or simply Pitt syndrome, it was later realized that these syndromes were part of the clinical spectrum of Wolf Hirschhorn [2] [3].
Wolf-Hirschhorn syndrome affects many parts of the body. The hallmark features of this syndrome are severe developmental delay, growth delay, intellectual disability, seizures, and characteristic facial features. This syndrome occurs in approximately 1/50 000 live births. WHS occurs in more females than males at a rate of 2:1 [2].
Description
A diagnosis of Wolf-Hirschhorn syndrome can be suspected clinically with the presence of distinctive facial features including:
- broad, flat, nasal bridge and high forehead (known as 'Greek Helmet Face')
- widely spaced, protruding eyes
- short philtrum (space between nose and upper lip)
- downturned mouth
- small chin
- ptosis
- ear malformations including pits or skin tags
- small head
- asymmetric facial features [2]
Individuals affected with Wolf-Hirschhorn syndrome also experience delayed growth and development. This is usually seen prenatally, with slow growth in utero , which is followed by low birth weight and size. Newborns may have low muscle tone, problems feeding, and trouble gaining weight (failure to thrive). Infants with WHS have underdeveloped muscles, especially of the lower limbs. They also experience developmental delays, with milestones like sitting, standing, and walking significantly delayed [2]. Most adults with WHS are shorter than average [1].
Most individuals with WHS are affected with some degree of intellectual disability, this can range from mild to severe. People with WHS have comparatively strong verbal and quantitative reasoning skills as well as higher socialization. Communication skills tend to be weaker in 65% of cases, with many individuals showing both expressive and receptive language delays [4]. Functionally, about 30% of individuals with WHS reach some level of autonomy with feeding, dressing, and household activities [2].
Seizures are a common feature of Wolf Hirschhorn syndrome, with presence in almost 100% of cases. Onset of seizures is usually in the first two years of life and are variable in presentation. Seizures are triggered by fever and often occur in clusters. Seizures can be managed with medication and usually stop at puberty in over 50% of individuals [2].
Over 50% of individuals experience skin changes (marbling or dry skin), skeletal anomalies (kyphosis, scoliosis, clubbed feet etc.), dental abnormalities (delayed eruption, missing teeth etc.) Less common features include birth defects, occurring in 25-50% of cases, such as: - heart defects (septal defects etc.) - eye or optic nerve defects (lens dislocation) - cleft lip or palate - conductive hearing loss - structural brain anomalies - genitourinary tract defects [2] [1]
WHS is a complex condition that can affect many different systems of the body. The severity of the condition and features vary from case to case. Some cases have reportedly mild symptoms such as characteristic facial features and mild intellectual disability, others report individuals with severe intellectual disability and multiple birth defects [5].
Genetics and Inheritance
Wolf Hirschhorn syndrome is caused by a deletion of genetic material on the short arm (also known as the 'p' arm) of chromosome 4. The size of the deletion varies between different individuals with WHS, some studies show that people with larger deletions tend to have more severe intellectual disability and physical abnormalities than with smaller deletions. The opposite of this has been seen in some cases [6][5].
-
Deletion of the short arm of chromosome 4 in a patient with Wolf-Hirschhorn syndrome
-
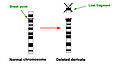 Diagram of a terminal chromosome deletion
Diagram of a terminal chromosome deletion

{kind=link}

The area of chromosome 4p that is deleted in WHS contains multiple genes that play important roles in early development. Lack of this genetic information leads to the complex condition seen in WHS. Three important genes in this area have been indentified in individuals with the key features of WHS: WHSC1, LETM1, MSX1. Some research shows that loss of WHSC1 gene is associated with facial appearance and developmental delay. Deletion of LETM1 is associated with seizures. MSX1 has been associated with dental abnormalities and cleft lip/palate. Further research is being done on this area of chromosome 4 to identify other genes that contribute to WHS [2][5].
90% of all cases of Wolf-Hirschhorn syndrome are not inherited, and result from random chromosome deletion (also known as de novo deletion) during the formation of gametes (egg or sperm) in development [2]. Other random chromosome rearrangements occurring on chromosome 4 can also cause WHS. De novo chromosomal changes occur in people with no family history of the condition.
In the remaining cases of WHS, individuals inherit a copy of chromosome 4 with a deleted segment. In these cases, one parent carries a rearrangement between chromosome 4 and another chromosome. This is called a balanced translocation and can be carried in individuals no signs or symptoms. The amount of genetic information is all present in individuals with balanced translocations so they usually have no resulting medical concerns. Translocations can become unbalanced, resulting in a gain or loss of genetic material when being passed on to the next generation. Inheriting an unbalanced translocation in the short arm of chromosome 4 can result in WHS.
Diagnosis and Treatment
Diagnosis of Wolf Hirschhorn syndrome can be made based on clinical features by a geneticist . Testing may also be offered to confirm a diagnosis. Depending on the size of the deletion this may require multiple genetic tests, all of which can be conducted using a blood sample. Deletions can be detected using traditional cytogenetics, by looking at a karyotype, a picture of all of the chromosomes in a cell that would show a missing part of chromosome 4. FISH techniques (fluorescent in situ hybridization) can be used to see deletions or rearrangements at higher resolution and in a specific area, in this case a section of chromosome 4p (4p16.3). Patients may also be offered chromosomal microarray to detect deletions at even greater resolution [2]. Identification of the size of the deletion is poor predictor or prognosis and severity in Wolf Hirschhorn syndrome [7].
There is no cure for Wolf Hirschhorn syndrome as it is caused by a chromosomal problem in all of the cells. Treatment is therefore geared towards treating the specific symptoms and needs of each affected individual. Treatment options include rehabilitation, speech/communication therapy, medical management of seizures, and GI tubing for feeding difficulty. The level of treatment required depends on the ability of the individual and specific symptoms of the condition present [4].
Either before, or after, a diagnosis is made, one can investigate the parts of the body affected by plotting growth, evaluating development, physical examinations, and assessing hearing, sight, cardiology, and the immune system. This can aid in leading to an accurate diagnosis or provide a guide for how to approach life-long management[2].
Genetic Counselling Issues
Genetic risk of WHS reoccurring in families depends on the origin of the deletion. 45% percent of cases of WHS are due to unbalanced translocation. These can be de novo or inherited from one parent with a balanced translocation [2].
If a couple has a child affected with Wolf Hirschhorn syndrome, it recommended that both parents be tested for a balanced rearrangement involving chromosome 4. If no rearrangement is found than the WHS in their child is de novo and there is minimal risk in future pregnancies of having another child with WHS. If a parent is found to be a carrier, there is an increased risk of having another affected child; this risk is dependent on the nature of the translocation.
If a parent is found to have a balanced translocation, his or her family members (including parents and siblings) are also at risk of carrying the rearrangement. Family members can be offered testing if they want information regarding their translocation status for future family planning.
The ideal time to discuss genetic risk (due to a previous affected child or family history), carrier status, and prenatal testing options is before pregnancy. Young adults who are carriers or at risk of being carriers can consider genetic counseling to discuss risks and reproductive options.
Prenatal diagnosis through invasive testing is available for families where one parent is known carrier of a 4p balanced translocation. Cells can be obtained by CVS or amniocentesis, and analyzed using cytogenetic methods (traditional karyotyping, FISH, or CMA depending on the rearrangement). Ultrasound findings can also raise suspicion of WHS, in fetuses presenting with limited growth and characteristic facial features. Couples should consider that results obtained cannot determine with certainty the severity of the condition in that pregnancy, there is considerable variability in the presentation of Wolf-Hirschhorn syndrome. They can however determine, if the fetus has a 4p deletion that is large enough to detect, or not. Pre-implantation genetic diagnosis may also be available for couples with a parental balanced translocation.
Resources

Individuals and families living with Wolf Hirschhorn Syndrome can feel alone and be faced with difficulties in everyday life. These support groups and resources allow families to contact each other and share experiences.
The Real Story about Wolf-Hirschhorn Syndrome
References
- ↑ 1.0 1.1 1.2 Online 'Mendelian Inheritance in Man' (OMIM) Wolf-Hirschhorn syndrome -194190
- ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 2.11 Battaglia A, Carey JC, South ST, et al. Wolf-Hirschhorn Syndrome. 2002 Apr 29 [Updated 2010 Jun 17]. In: Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1183/
- ↑ Kant, S. G., Van Haeringen, A., Bakker, E. et al. (1997) Pitt-Rogers-Danks syndrome and Wolf-Hirschhorn syndrome are caused by a deletion in the same region on chromosome 4p16.3. J. Med. Genet. 34: 569-572
- ↑ 4.0 4.1 http://wolfhirschhorn.org/about-wolf-hirschhorn-syndrome/
- ↑ 5.0 5.1 5.2 Bergemann, A.D., Cole, F., and K. Hirschhorn. (2005) The etiology of Wolf-Hirschhorn syndrome. Trends in Genetics. 21(3):188-95.
- ↑ Zollino, M., Murdolo, M., Marangi G., et al. (2008). On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C Semin Med Genet.148C:257–69
- ↑ Meloni A, Shepard RR, Battaglia A et al. (2000). Wolf-Hirschhorn syndrome: correlation between cytogenetics, FISH, and severity of disease. Am J Hum Genet. 67:149